Clustering
This page describes how to cluster a gene by sample table based on the samples or the genes, and how to evaluate the results. Code discussed in the paper can be found in the stuartlab clustermethods github repo
1. Determine number of clusters
Most clustering methods require a preset number of clusters. Unfortunately there's no way to automatically determine the optimum number of clusters so you will have to do some preliminary analysis.
The best way is to visualize your data using a dimensionality reduction method such as t-SNE or TumorMap. Simply input your batch corrected gene by sample table and look at the resulting figure.
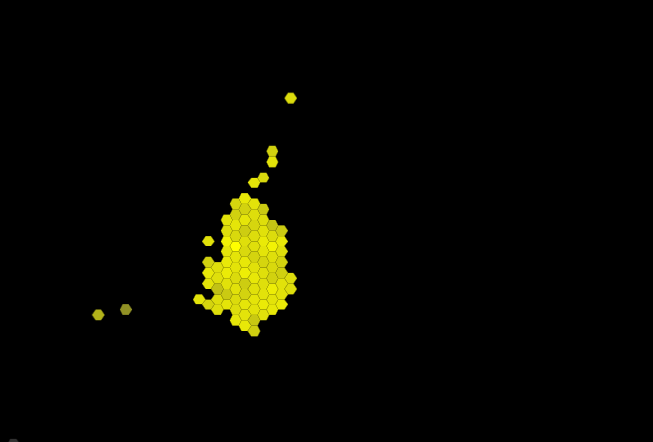

If your data comes out as a single blob on either of these methods, you likely won't be able to get multiple clusters from the clustering algorithms. Of course you can request three clusters and the programs will give you something, but it's likely meaningless and will differ between methods.
2. Log2(TPM/FPKM)+1
If you haven't already done so, convert your FPKM input to log2(TPM)+1
3. Clustering methods
The program clustermethods.py takes your gene by sample table and your required number of clusters. First, it removes the genes with low expression and then it selects the top 1000 most variable genes. Using fewer genes tends to lead to better outcomes. It then runs the following clustering methods:
- Spectral biclustering. This method clusters genes and samples at the same time.
- Hierarchical clustering with a correlation based distance matrix and average linkage
- Hierarchical clustering with a correlation based distance matrix and complete linkage
- Hierarchical clustering with ward linkage (creates a euclidean distance matrix)
- Hierarchical clustering with spearman rank and average linkage
- Hierarchical clustering with spearman rank and complete linkage
- Kmeans
- Kmedoids
If you want to cluster genes instead of samples, set the --genes flag. The program will now run on the filtered gene set, without selecting the 1000 most variable genes.
4. Scoring
For each method, the program caculates the silhouette score and for the hierarchical methods it also calculates the cophenetic index (which can only be calculated on dendrograms). These scores say something about how well the method fits the data, but note that the cophenetic index will not change if you alter the cluster number. This is because it gets calculated on the full tree, not on the number of clusters you request (which is a step that happens afterwards).
5. Output
To help you visualize the results, the program outputs a Tumormap-ready input feature map with labels for each clustering method. You can also opt to output the reduced gene set that was used for clustering and use this as Tumormap's layout input or as input to another iteration to the clustermethods.py program. In both programs this will increase upload and calculation speed.
You can opt to get a list of grouped samples. These are samples that end up in the same cluster in all the methods used in the program.
6. Making changes
The clustermethod.py code and the associated libraries are well commented and you should feel free to check out a copy and play with it.
Most of the clustering methods used are from Python's scikit learn package, which has extensive documentation and example code. You should be able to add additional methods from this package, or from scipy clustering.